
Well, we’re finally here. The last blog post. The last of the unconquered waters of AP Chem. Right before sailing through AP Exam Reef and docking into the harbor one last time.
Before Spring Break, we had finished off the topic of electrochemistry – the Nernst equation to determine the E of the cell in nonstandard conditions and the second type of power cell, the electrolytic cell. First of all, the Nernst equation is actually a manipulation of the equation used to determine the ∆G of a reaction in nonstandard equations. Basically, it is said…
-nFE = ∆G ; n= moles of electrons, F is Faraday’s Constant
So replacing ∆G with -nFE and cancelling out the -nF yields the Nernst equation:
E = E^o -RT/nF ln Q
The key all lies within the natural log of Q. Basically, Q is the K of the system, which accounts how the different concentrations wouldaffect the spontaneity and E of the cell. 
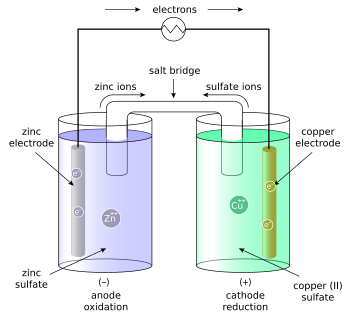
On the opposite spectrum of spontaneity is the electrolytic cell, which is not driven by free energy whatsoever. Rather, the cell works when a current is run through (a.k.a. energy is put into the system). Because of this unique property, there is no need for a separation of chambers, but only a controllable power source. Typically with electrolytic cells, electrolysis is performed, where it’s possible for the water that holds the substances to take part in the reactions. For example, around the anode of the cell, either the electrode substance or the water can be oxidized – it all depends on which half-reaction has the more positive E^o, and the same holds true for reduction at the cathode end.
One of the biggest applications of electrolysis is the process of electroplating – basically running a current through a vat of water in order to oxidize the plating metal into solution and reducing it around the desired object. Typically, this occurs with gold and silver, but it’s possible for many substances to do this – the week before Spring Break’s lab focused on this aspect with copper.
As depicted above, we were electroplating copper from one strip to the other, depending on which one was the anode and which one was the cathode. This lab was particularly fun because 1) it was essentially the opposite of the first lab we ever did in AP Chem at the beginning of the year, and 2) we were allowed to copper plate some of our possessions, like dull pennies (returning the sheen) or non-copper plated coins. In the end, we were basically transferring copper from one plate to the other.
As for any hardships on the Chem journey, I still often more than not get confused about flipping signs and whatnot. Do I flip the E and add, or leave it and subtract? Should I do both? Honestly, just thinking about confuses me albeit less intensely. Some usage of the Nernst equation also gave me trouble too, but that was mostly from forgetting to change some units here or there, or just plain old forgetting to include the temperature in the equation. Being a chemist sure has its moments, huh?
Well, it’s sad to see this blog reach its last post for the known future. Looking through the posts is like going down memory lane – I can’t believe it’s already been an entire school year. College is on the horizon, and I’m just speeding closer and closer with broken brakes. All things considered though, it’s been a hell of a ride. Now, all that there’s left for me to do is to ace the AP Chem test and graduate. I might as well increase the entropy of the universe while I’m at it. So, until next time!
Ensign Hung, signing off!











 A week in AP Chemistry would be pretty uneventful without one of its trademark labs. Lucky for us, we did a lab that had us determine the Ka of 4 unknown weak acids. They were all in the solid state, so we would have to dissolve the acid ourselves in 50 mL of distilled water. From there, we would split the solution into two 25 mL samples, and titrate one of the samples with NaOH base until the indicator inside of the solution turns a faint fuchsia to indicate the solution’s neutralized status. Then, we reintroduce the two samples in a beaker and find the pH.
A week in AP Chemistry would be pretty uneventful without one of its trademark labs. Lucky for us, we did a lab that had us determine the Ka of 4 unknown weak acids. They were all in the solid state, so we would have to dissolve the acid ourselves in 50 mL of distilled water. From there, we would split the solution into two 25 mL samples, and titrate one of the samples with NaOH base until the indicator inside of the solution turns a faint fuchsia to indicate the solution’s neutralized status. Then, we reintroduce the two samples in a beaker and find the pH.










{kind=link}